 |

|
|
|
|
 Back to 1997 4th Quarter Table of Contents
Back to 1997 4th Quarter Table of Contents
Abstract Children presenting with Autistic Spectrum Disorder have challenged medical science, yet prolific research has not answered the cause, appropriate testing or treatment of these patients. To determine the etiology of children with autistic features we investigated metabolic aberrations through profiling blood chemistry, organic and amino acids, and red cell membrane fatty acids. Our findings of the red cell membrane analysis of 50 subjects reveal characteristic elevation of the very long chain fatty acids (VLCFAs) above C22 as Nervonic (C24:1w9), Lignoceric (C24:0), Docosapentaenoic (C22:5w3) and Docosahexaenoic (C22:6w3) indicating peroxisomal involvement. Peroxisomal disorders are characterized by an accumulation in tissue and body fluids of metabolites that normally are degraded in the peroxisome including saturated and unsaturated VLCFAs and the branched chain fatty acids, pristanic and phytanic. Peroxisomes are pivotal in the biotransformation of endogenous compounds in lipid metabolism as fatty acids, steroids, prostaglandins, the formation of myelin impacting the immune, endocrine and central nervous systems in addition to the detoxification of exogenous compounds and xenobiotics. The accumulation of VLCFAs are indicative of defects in peroxisomal ßoxidation which may explain the deleterious effects to the brain, endocrine and immune systems and hepatic cytochrome P450 derangement characteristic in autistic spectrum disorder. Introduction Profound neurodegenerative conditions observed in autistic spectrum disorder (ASD) with or without seizures in
pediatric patients can be attenuated with metabolic intervention if unique aberrations in red cell fatty acid analysis are carefully profiled through a medical data management system and attenuated specific to unique fatty acid disturbance. Our theoretical model suggests that because children with ASD present with a wide variation of soft signs of metabolic derangement, many aspects of an individual’s biochemistry must be evaluated and metabolically modeled. Our interpretation of findings are based on this metabolic model with the evaluation of energy generation and transfer in the forefront due to the aberrant findings in our study participants which can be traced to a secondary method of energy generation riveted within altered fatty acid metabolism ultimately impacting the peroxisomal organelles. A new frontier in medicine has awakened in the exploration of three dimensional metabolics through red cell fatty acid analysis opening a therapeutic window in time—a two month view—into cell membrane dynamics. The bilipid layer surrounding every cell and organelle is far more than a protective shield; it is the very essence of life, with vibrant electrical activity providing gates in and out–a bewildering array of receptors for opening and closing the bilipid layer for ingress and egress–a dazzling assembly of signaling devices that literally control and signal all activities of membrane traffic inside and out. Red cell fatty analysis leads the clinician into a wide realm of metabolic strategies to influence the health of the patient. Lipids evolve into hormones, immune components, leukotrienes, prostaglandins, cytokines, and myelin–there is virtually no system of the body that does not require attenuation of specific fatty acid substrates and coenzymes to maintain health and repair of body tissues. The turmoil over which specific lipid substrates to administer for therapeutic application to fulfill individual need has always been a loaded question that has led physicians to an awkward gunshot approach (just add flax oil to the diet) that has often failed us therapeutically. It is prudent that the specific essential fatty acid needs of patients be addressed in a scientific manner so that favorable clinical outcomes might be reached. Clinical application of research data often has left physicians frustrated on timely introduction of new testing methods and metabolic support directly to patients. Children with ASD present an even greater frustration due to the complexity of their illness. Clinical lab results are redefined through Carbon Based computation of raw data offering the physician metabolic strategies that may be applied immediately. In the case of two year old Alex, his parents were gravely concerned over their child’s diagnosis of autism. His parents and doctor were interested in biochemical intervention, but were unsure how to assess the disturbed metabolism and immune insufficiency. We suggested initial labs of a Chemistry Panel-28 including a CO2, Complete Blood Count with differential, and a Red Cell Membrane Fatty Acid Analysis from Kennedy Krieger Institute Peroxisomal Diseases Lab. Alex’s red cell membrane fatty acid test results revealed strident elevation of Docosapentaenoic w3, Docosahexaenoic, Lignoceric, Arachidonic and Nervonic acids (Table 1, p. 209). Alex’s blood chemistry was also sharply altered (view Alex’s complete blood chemistry before and after nutrient therapy Table 2, p. 210, Table 3, p. 211) with derangement in his hematology, mineral balance, nitrogen retention, electrolyte and immune parameters. Alex’s parents and doctor were able to begin immediate administration of appropriate nutrient therapy and directed nutriture with receipt of Alex’s lab results run through the Carbon Based system. The child required substantial digestive support, mineral repletion, oral electrolytes, cofactors, and targeted lipids. Within three months of metabolic intervention Alex had made remarkable strides in speech (absence of speech to 48 words), eye contact, appropriate play, social interaction and gross motor skills. Eight months of targeted nutrient support through blood chemistry and lipid evaluation yielded complete cessation of autistic features (see comparison report Table 4, p. 212). Methods Collection of data involved the aquisition of pertinent blood and urine specimens for analysis of blood chemistry, hematology, organic acids, amino acids and red cell membrane fatty acids along with complete medical records and an extensive 35 page questionnaire of the child’s history including pictures and video footage of child’s developmental delay and medical condition. Blood chemistry and hematology specimens were sent to LabCorp in Reno, Nevada, Urine and plasma organic acids and amino acids (mass spectrometry) to Saint Louis University Metabolic Screening Lab, and RBC fatty acid analysis (gas chromatography) to Kennedy Krieger Institute Peroxisomal Diseases Laboratory in Baltimore, Maryland. In depth computational data analysis and graphical representation from Carbon Based Corporation in Incline Village, Nevada was utilized to evaluate biochemical laboratory results and metabolic interactions of individual patients, a comparative model of the chemistries, drug interactions, and disease patterns. This nexus of modern computational technique with raw data potentiates an exceptional scientific tool enabling medical data to be more fully utilized. Results Elongation of very long chain fatty acids in red blood cells as Nervonic (C24:1 w9) and Lignoceric (C24:0) acids indicative of peroxisomal involvement was the most Table 1. Red cell fatty acid membrane analysis of Alex’s blood lipids profile.  Table 2. Alex’s autism. Initial blood chemistry of Alex, age 2, before targeted nutrient therapy. 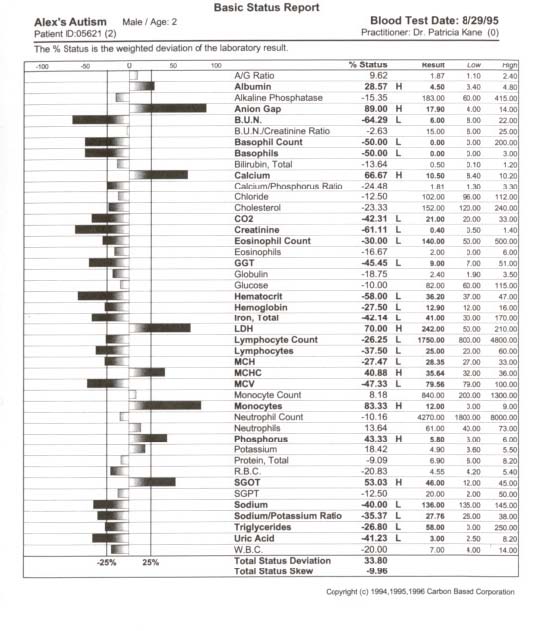 Table 3. Alex’s autism resolved. Post treatment blood chemistry of Alex, age 3, after targeted nutrient therapy. 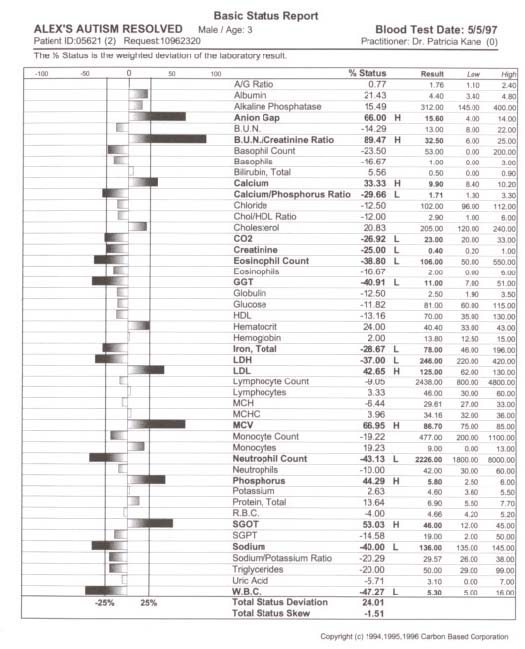 Table 4. Comparison report of blood profile of Alex before and after targeted nutrient therapy. The arrow’s length is proportional to change. Left is increased; right is decreased; White is an improvement; black is a decline. 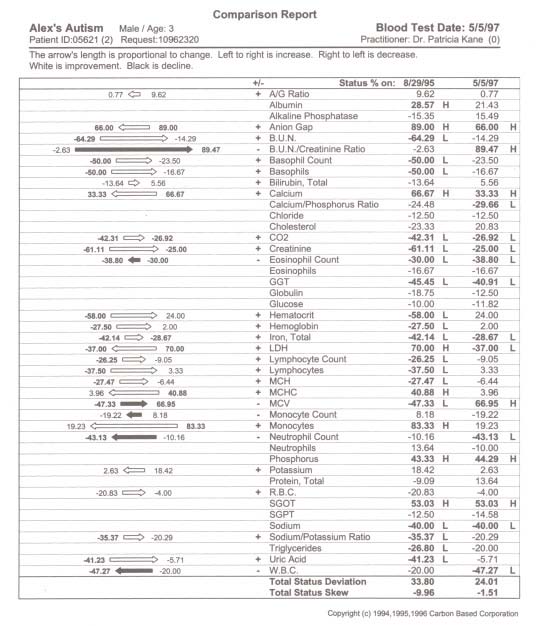 prominent and consistent feature in every subject tested, with 39 out of 50 subjects also exhibiting elevation of Docosapentaenoic (C22:5w3) and Docosahexaenoic (C22:6w3), in 41 of 50 subjects Arachidonic (C20:4w6) and 20 of 50 subjects elevation of Eicosapentaenoic (C20:5w3). Depression of fatty acids in red blood cells included 30 out of 50 subjects (primarily subjects with profound autistic features) with supression of Eicosapentaenoic (C20:5w3) and 50 out of 50 subjects with suppression of Gamma Linolenic (C18:3w6) and Dihomogamma Linolenic (C20:3w6). Treatment Subjects with elevation of VLCFAs required detailed manipulation of their individual biochemistry to address fully the buildup of VLCFAs. Children with autistic spectrum disorder were approached initially with stabilization of membrane traffic through examination of their serum electrolytes and subsequent administration of an oral balanced hypertonic electrolyte solution in enteral or oral feeds. Marked disruption in ionic homeostasis pH, anion gap, cellular osmolarity, and the decline of electrolyte gradients across the cell membrane was observed as these patients have grave difficulty balancing the dynamic process of their chemical buffering systems to maintain a constant serum pH. Often there was involvement of all three systems: Carbonic acid-Bicarbonate buffering; Phosphate buffering; and Protein buffering systems. Children with intractable seizure disorders, autistic syndromes and brain injury have improved cognitively when receiving individualized IV electrolyte solutions during hospitalization for illness (as observed by Kane) whereby positive outcomes also were achieved with balanced 9 mEq oral electrolyte solutions. Metabolic intervention also was directed towards lipid manipulation by competitive inhibition,1 inclusion of the lignan sesamin2 and curcumin3 with low CHO diet to suppress insulin4-8 and block delta 5 desaturase synthesis, the administration of coenzymes such as potassium permanganate, riboflavin-5-phosphate,9-10 selenium and hormones11 as dehydroepiandrosterone,12 pregnenolone,13-15 and thyroid16 to stimulate betaoxidation, nutritional adjuncts to stabilize lipids in the cell membrane17-25 such as ginger, oral balanced electrolytes and alteration of the diet to increase nutrient density, avoidance of VLCFAs in food intake, and supplementation of depressed fatty acids26 (determined from red cell testing) such as evening primrose oil27 to address suppression of GLA/DGLA and long chain fatty alcohols to stimulate plasmalogen synthesis.28,29 Conclusions Derangement of lipid metabolism primarily of peroxisomal ßoxidation was observed in children with seizure disorders,30 autism,30 ASD with enlarged neurons,31 and hypoxic brain damage32 with sharp elevation of VLCFAs with elevation of Lignoceric, Nervonic, Docosahexanoic, and depression of Hexacosanoic. Conversely, boys with X-Linked Adrenoleukodystrophy exhibit elevation of serum and red cell Lignoceric and Hexacosanoic acids33 with depression of Docosahexaenoic34 and Nervonic acids.35 Peroxisomal disorders are characterized by an accumulation in tissue and body fluids of metabolites that normally are degraded in the peroxisome including saturated and unsaturated VLCFA,33, 36-41 branched chain fatty acids, pipecolic, phytanic and pristanic acids, and bile acid intermediates dihydroxy- and trihydroxycoprostanic acid, (DHCA and THCA respectively). Peroxisomal disorders with a clinical onset of four years of age may present as behavioral changes, intellectual deterioration, and visual impairment. Accumulation of VLCFAs42 are associated with a deficiency of the fatty acyl-CoA oxidase (AOX), the enzyme that catalyses the first step in ßoxidation. A prerequisite for ßoxidation is the activation of fatty acids to their Co-A derivatives.43-45 Peroxisomes and mitochondria degrade saturated and unsaturated fatty acids via similar reactions with the same acyl-CoA intermediates formed, but there are distinct differences between the two systems.46-48 Very long chain acyl-CoA synthetase activity is present in the endoplasmic reticulum and proposed to be in the peroxisomes, but not in the mitochondria.49-52 Long chain fatty acids are preferentially oxidized in the mitochondria,53-55 but VLCFAs are oxidized predominantly, if not exclusively in some tissues by peroxisomes.56,57 Compared to long chain fatty acids (in mitochondria), the VLCFAs are only slowly oxidized by isolated peroxisomes. VLCFAs enter the peroxisome58 and are shortened to long chain fatty acids, transferred back to the mitochondria,59 then further oxidized in mitochondria, esterified in the endoplasmic reticulum or perhaps partly in the peroxisome itself. The accumulation of VLCFA may constitute a minor part of overall fatty acids, but peroxisome deficiency disorders are deleterious to the brain and CNS.33,60 Autism and seizure disorders may mimic pseudo-neonatal adreno-leukodys-trophy61,62 as a peroxisomal disorder with enlarged peroxisomes30,31 and a specific deficiency of acyl-CoA oxidase.63,64 Peroxisomes, present in virtually all cells, but most prevalent in the liver and kidney play a critical role of cellular lipid metabolism in the biosynthesis of fatty acids via ßoxidation65-69 involving physiologically important substrates for VLCFA, dicarboxylic fatty acids, prostaglandins, thromboxanes, leukotrienes, pristanic acid, DHCA, THCA, and xenobiotics.70,71 Children with autistic spectrum disorder often present with complex xenobiotics involving disturbances in the cytochrome p450 superfamily which parallels disturbances in peroxisomal function.72-82 The cytochrome p450s are responsible for the biotransformation of endogenous compounds including fatty acids, steroids, prostaglandins, leukotrienes and vitamins as well as the detoxification of exogenous compounds resulting in substantial alterations of p450s as xenobiotics may turn off or greatly reduce the expression of constitutive isoenzymes. The overzealous administration of tocopherols may actually worsen the condition of autism as vitamin E is a potent antioxidant that supresses beta-oxidation of the very long chain fatty acids. Elevation of Docosahexaenoic acid (DHA)83-85 in red cell fatty acid analysis found in many of our subjects with autistic spectrum disorder may involve the cytochrome p450 enzyme Nitric Oxide Synthase (NOS) and Nitric Oxide (NO) formation in that supplementation of DHA augments NO generation.86 Nitric oxide inhibits beta oxidation of lipids and thereby stimulates the buildup of VLCFAs. Nitric oxide is the smallest biologic product of human cells, critically involved in the modulation of cerebral blood flow, blood pressure, electrolytic balance, platelet adhesion, hormonal release, insulin secretion, synaptic plasticity, immunity, neurotransmission, neuromodulation, gastrointestinal and hepatic function. Cerebral injury or hypoxia mediates an excess of the neurotransmitter glutamate which acts at the NMDA subtype receptor to open Ca2+ channels creating an increase in NO release with inducible NOS activity (there are three isoforms of NOS, inducible NOS is Ca dependent) detected in immune cells within the brain and elevation of cGMP levels.87 The synthesis of NO by neurons has finite regulation and NO’s reactions vary widely with pH. Nitric oxide has a high affinity for hemoglobin and NO is rapidly inactivated by binding to hemoglobin in that its physiological actions remain localized to the site of its generation and its actions are in general rapidly terminated. This phenomenon may explain why many subjects in our study exhibited sharp elevation of hemoglobin, iron and RBCs yet presented with a “china doll complexion” (aptly described by Carl Pfeiffer). Nitric oxide activity can be prolonged by forming nitrothiols with serum albumin88 acting as an NO carrier. Thus the consistent sharp elevation of albumin in our subjects may be understood in that nitrothiols may act as a store, prolonging the actions of released NO or as an additional “biological sink” for NO which regulates the concentration of free NO. While Arachidonic acid induces cyclooxygenase (COX) activity, nitric oxide specifically inhibits COX-2. As nitric oxide binds to heme, therapeutic modalities may be applied to reach COX-2 expression whereby expression of COX-2 may dictate the balance between TH1 and and TH2 cells presently being investigated in ASD. Eicosapentaenoic acid (EPA) down-regulates tumor necrosis factor having profound immunomodulatory properties. Perhaps the pivotal issue as to why children with pervasive developmental delay and autism do not respond appropriately to administration of marine oils is that they are unable to beta oxidize DHA into EPA or convert EPA into series three prostaglandins. Our findings indicate that the electrolyte disturbances (primarily involving elevation of calcium with depression of carbon dioxide, elevation or suppression of sodium and potassium), altered hematology, compromised nitrogen status (low creatinine, elevated albumin) and elevation of red cell fatty acids Docosahexaenoic, Docosapentaenoic w3, Arachidonic, Nervonic and Lignoceric may serve as crucial markers in neurodegenerative conditions leading to clarification of the etiology of autistic syndromes and complex seizure disorders by approaching metabolic derangement through targeted lipids, long chain fatty alcohols, pregnenolone (only after appropriate lipid therapies have been administered), oral electrolytes, coenzymes (vitamins and minerals as cofactors), and dietary manipulation (elemental dietary measures). Autism is a systemic disorder that has plagued yet intrigued us for decades. To unravel autism inspires us to view medical disorders and disease from both a microcosmic and macrocosmic biochemical perspective. By capturing cell membrane dynamics we now have an individualized, targeted therapeutic approach to ease the entropy of disturbed metabolism. References
riboflavin-responsive myopathy with combined multiple acylcoenzyme a dehydrogenase and respiratory chain deficiency. Neurology, 1994: 44; 2153-2158.
49.Rapoport SI: In vivo labeling of brain phospholipids by long-chain fatty acids: relation to turnover and function. Lipids 1996: 31; S97-S101. 50.Watkins PA, McGuinness MC, Raymond GV, Hicks BA, Sisk JM, Moser AB, Moser HW: Distinction between peroxisomal bifunctional enzyme acyl-CoA oxidase deficiencies. Annals of Neurology 1995: 38/3; 473-477. 51.Tserng KY, Chen LS, Jin SJ: Comparison of metabolic fluxes of cis-5-enoyl-CoA and saturated Acyl-CoA through the ßoxidation pathway. Biochem J, 1995: 307: 23-28.
54.Lazo O, Contreras M, Singh I: Topographical localization of peroxisomal Acyl-CoA ligases: differential localization of palmitoyl-CoA and lignoceroyl-CoA ligases. Biochemistry, 1990: 29: 3981-3986.
62.Kyllerman M, Blomstrand S, Mansson JE, Conradi NG, Hindmarsh T: Central nervous system malformations and white matter changes in pseudo-neonatal adrenoleukodystrophy. Neuropediatrics, 1990: 21; 199-201. 63.Araki E, Kobayashi T, Kohtake N, Goto I, Hashimoto: A riboflavin-responsive lipid storage myopathy due to multiple acyl CoA dehy-drogenase deficiency: an adult case. J Neurolog Sci, 1994; 126: 202-205. 64.VanHove GF, Van Veldhoven PP, Fransen M, Denis S, Wanders RJA, Mannaerts GP: The CoA-esters of 2-methyl-branched chain fatty acids and of the bile acid intermediates Di- and trihydroxycoprostanoic acids are oxidized by one single Peroxisomal Branched Chain Acyl-CoA oxidase in human liver and kidney. J Biol Chem, 1993: 268; 10335-10344.
74.Guengerich FP: Reactions and significance of cytochrome p450 enzymes. J Biol Chem, 1991: 266; 10019-10023. 75.Porter TD, Coon MJ: Cytochrome p450 multiplicity: isoforms, substrates and catalytic and regulatory mechanisms. J Biol Chem, 1991; 266; 13469-13472. 76.Roels F, Espeel M, Poggi F, Mandel H, Van Maldergem L, Saudubray JM: Human liver pathology in peroxisomal diseases: a review including novel data. Biochimie, 1993: 75: 281-292. 77. Sharma R, Lake BG, Foster J, Gibson GG: Microsomal cytochrome p452 induction and peroxisomal proliferation by hypolipidaemic agents in rat liver: a mechanistic inter-relationship. Biochem Pharm, 1988; 37; 1193-1201.
80.Ram PA, Waxman DJ: DHEA 3 ßsulfate is an endogenous activator of the peroxisome-proliferation pathway: induction of cytochrome p450 4a and acylco oxidase mRNAs in primary rat hepatocyte culture and inhibitory effects of ca++ channel blockers. Biochem J, 1994: 301; 753-758. 81.Luers G, Beier K, Hashimoto T, Fahimi HD, Volki A: Biogenesis of peroxisomes: sequential bio-synthesis of the membrane and matrix proteins in the course of hepatic regeneration. Euro J Cell Biol, 1990: 52; 175-184.
85.Boulanger C, Schini VB, Hendrickson H, Vanhoutte PM: Chronic exposure of cultured cells to eicosapentaenoic acid potentiates the release of endothelium-derived relaxing factor(s). Brit J Pharmacol, 1990: 99; 176-180.
|

This website is managed by Riordan Clinic
A Non-profit 501(c)(3) Medical, Research and Educational Organization
3100 North Hillside Avenue, Wichita, KS 67219 USA
Phone: 316-682-3100; Fax: 316-682-5054
© (Riordan Clinic) 2004 - 2024c
Information on Orthomolecular.org is provided for educational purposes only. It is not intended as medical advice.
Consult your orthomolecular health care professional for individual guidance on specific health problems.
 Download The Full Text Article in (PDF)
Download The Full Text Article in (PDF)